一 : wwwhxtborg 《化学通报》在线预览版 微流控芯片中的流体驱动和
http://www.hxtb.org 《化学通报》在线预览版
微流控芯片中的流体驱动和控制方式
徐溢1,2*,陆嘉莉,胡小国,任峰 111
(1重庆大学化学化工学院,2重庆大学微系统研究中心, 重庆 400030)
摘要 随着微流控芯片分析技术的发展,微流控芯片中的关键技术之一流体的驱动和控制技术逐步成为人们关注和研究的热点。[www.61k.com)本文主要综述了微流控芯片上的压力驱动、电驱动和其他驱动方式,着重介绍了电驱动方式,并讨论了各种驱动方式的优缺点以及发展前景。
关键词 微流控芯片 微泵 电渗流 阵列电极 压力驱动
Driving and Controlling Techniques of Microfluid in Microfluidic Analytical System
Xu Yi1,2, Lu Jiali1, Hu Xiaoguo1, Ren Feng1
(1College of Chemistry and Chemical Engineer, 2Mcrosystem center , Chongqing university, Chongqing 400030)
Abstract With the development of the microfluidic analytical system, microfluidic driving and controlling technique, as one of its key techniques, is concerned and researched by relative experts. According to its principle, pressure driving, electric driving and other driving techniques, especially the electric driving techniques were reviewed in this paper. Their advances and prospective are discussed respectively.
Keywords Microfluidic chip, Electric driven, Electroosmotic flow, Microarray electrode, Pressure driven
微全分析系统(μ-TAS)、芯片实验室或者集成化学实验室是1990年提出的分析化学的新领域,基于芯片结构的μ-TAS中,依据芯片结构及工作机理又可分为微流控芯片和微阵列(生物)芯片,它们都依托于微电机加工技术,又都主要服务于生命科学,但前者以微通道网络微结构特征,后者则以微探针阵列微结构特征。微流控芯片分析系统具有广泛的适用性及应用前景,随着MEMS微制作技术的迅速发展,相关研究和技术在20世纪90年代中期迅速崛起,备受人们的关注。微流控芯片分析系统主要通过对微通道内流体的操控,在芯片系统中完成包括采样、稀释、加试样、反应、分离、分析检测等功能。研究与微通道相适应的微流体驱动技术是实现微流体控制的前提和基础,微流体的驱动与控制和宏观流体的驱动与控制有很大的不同,这主要是由于尺度减小,流体的流动特性发生了变化。这种流动特性的变化使得宏观流体驱动与控制技术在微流体中的简单移植往往不成功或者效果不好,微流体的驱动与控制技术更为复杂化和多样化。
微流体的驱动和控制技术种类很多,采用的原理和形式不尽相同,可分为压力驱动、电驱动、热驱动、表面张力驱动、离心力驱动等。本文按照原理分类,重点综述在微流控芯片上实施压力驱动、电驱动方式及其它几种驱动方式的研究现状和特色,讨论了各种驱动方式的现优缺点及发展前景。
1. 微流控芯片上的流体驱动和控制方式
1.1压力驱动和控制方式
微流体的压力驱动和控制与宏观流体的原理相似,都是依靠入口、出口和腔体内部的相对压差来驱动流体,利用机械阀实现了流动控制。目前,利用压力驱动和控制微流体归纳起来有两种方法。一种利用外国家自然科学基金(20675089)(90307015)、科技部863计划项目(2006AA04Z345)和国际科技合作项目(2006DFA13510)、重庆市自然科学基金重点项目(CSTC, 2006BA4012)和(CSTC-2006BB7184)资助。
徐溢 wwwhxtborg 《化学通报》在线预览版 微流控芯片中的流体驱动和
部的宏观泵或注射器与微流体管道耦合,通过前者的推动力驱动流体在微管道中流动,流体冲开管道中的阀门被释放出,这种方法简单、容易实现、成本低,而且已经商业化,但不易小型化是它的一个主要缺点;另一种是采用微机械技术制作的微泵来提供压力。(www.61k.com]
Smits[1]最先报道了有关微机械泵的研究,第一台基于薄膜的往复运动驱动流体的微机械泵是在1988年由荷兰Twente大学的Zijtel等[2]提出的。该泵由一个压电陶瓷驱动的泵膜以及入口阀和出口阀组成,薄膜泵的主要问题是输入和输出止回阀的泄漏。1993年Stemme等提出一种无阀微机械泵[3,4],其中薄膜式微泵的止回阀被扩散器/喷嘴单元代替,扩散器/喷嘴单元在两个方向上的流阻不同。1995年,Zengerled等[5]提出一种可以前向和逆向两个方向驱动流体的微机械泵,与以往的阀设计的不同在于它是利用阀与驱动流体的压差响应之间的相移来实现流体的双向驱动。1996年Stehr等[6]提出了VAMP(valve and micropump)的装置,其既可以当作主动阀使用,也可以当作可两个方向工作的微泵使用。后来又出现了压电驱动微泵、热驱动微泵[7]等多种不同驱动薄膜振动方式的微泵。压电驱动微型泵是通过晶体的压电特性来驱动薄膜振动, 以达到输送工作液体的目的。Nguyen等[8]研制的压电驱动微型泵是由有机玻璃和SU-8光刻胶[9]组成,与硅材料相比,光刻胶较低的弹性常数使压电驱动电压低几个数量级,即几十伏的驱动电压就能满足泵的正常工作。
微泵开始是作为微流控系统外的一个独立器件,现在逐渐发展为在芯片上集成微泵以形成一个连续的操作系统。芯片上制作微泵和微阀虽然从芯片系统集成角度是很好的理念,有利于形成微型化、集成化、便携化的微流控芯片分析系统,但是传统的有阀微泵由于其结构复杂、工艺繁琐,因此无论是从技术或者成本上看都很难集成到微流控芯片上去,而微型无阀泵由于其简单的平面结构特征使得集成有很好的前景,因而,现在关于微泵的研究趋向于把无阀微泵集成到微流控芯片上。
1.2电驱动和控制
在微流控芯片分析系统中,电驱动还是最常用和最有效的驱动方式之一。它通常是在储液池的两端放置外电极,通过在电极上施加电压,在溶液中形成驱动电场来实现微管道中的液体的驱动,这也是目前芯片电泳分析系统的主流驱动方式。近年来,由于微机械加工技术的发展,使得芯片上集成电极成为可能,出现了在芯片上利用集成阵列电极来施加电场,实现微流体驱动的方式。阵列电极的方式主要应用在介电电泳芯片、低电压电泳芯片和交流电渗泵等方面。
61阅读提醒您本文地址:
1.2.1芯片外置电极方式的电驱动 在微流控芯片分析系统中,利用电渗流来驱动流体在微管道中流动,是一类较为成熟的方法,也是目前最成功的微流体驱动和控制方法之一。电渗驱动属于致动力直接作用于流体的驱动方式,其原理是利用微通道表面存在的固定电荷进行驱动。以玻璃基质微芯片为例,在中性或碱性pH下,玻璃通道表面带负电荷,液流中与其相邻的部分形成沿通道壁的带正电荷的截面―双电层由此产生,在通道两端施加高压,带正电荷的界面在电场作用下产生迁移,继而带动通道内界面包裹的液流产生电渗流―液体的流动。双电层厚度通常只有数十纳米,因此,电渗泵只能在极小的微通道内工作。
Harrison等[10]用电渗流来驱动流体,成功实现了微芯片上的电泳分离实验,这种技术经过不断完善,已被广泛应用于生物芯片等微型化学分析系统中样品的传输和控制;Harrison等[11]在微机械技术制作的宽度为20μm的玻璃管道中获得的电渗流流速达到1cm/s,典型的电渗驱动技术的流速在10nL/s~0.1μL/s之间。Schasfoort等[12]利用50V的电压在垂直微管道的方向上产生1.5MV/cm的电势差,利用该电势差可实现对电渗流大小和方向的控制,利用两个电渗流薄膜场效应晶体管(FlowFET),甚至可以逆转单管道中电渗流的方向。Banchi 等[13]通过对T型微尺度下的电渗驱动的模拟,对电渗流进行了理论分析。Laurie 等[14]
徐溢 wwwhxtborg 《化学通报》在线预览版 微流控芯片中的流体驱动和
用电流监测法测定了几种材料微芯片上的电渗流,得到共聚多醚的电渗流最大值达4.3×10-4cm2V-1s-1,这个值与之前报道过的熔硅毛细管的电渗流值相近。(www.61k.com]用压印方法(wire-imprinting method)制作的聚苯乙烯材料、丙烯酸材料以及用激光刻蚀方法制作的聚苯乙烯材料的微芯片的电渗流值分别为1.8、2.5、4.5×10-4 cm2V-1s-1。流速-电场强度的线性范围在100Vcm-1到500 Vcm-1之间,而在这个范围内,散热性能较好,能够满足一般电泳分离的需要。Pittman等[15]对用电流监测法进行玻璃微芯片的电渗流测定中的电渗流动力学进行了实验研究。Sun等[16]制作的用于细胞分类的微流控芯片也是利用电渗来驱动。
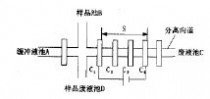
由于采用电渗驱动和压力驱动时分别存在细胞引入困难和细胞流速难以控制的问题,姚波等[17]采用静压力与电渗流模式相结合的方式,进行细胞的驱动和检测,如图1跟图2。首先利用页面高度形成的静压力差驱动细胞进入芯片通道,当细胞运动到十字交叉处,受电场力和静压力两种作用继续迁移而通过检测器。静压力较小,主要是使细胞较容易从储液池进入微管道,而电场力主要是将细胞输送到检测器的主要动力。


电渗驱动相对于其它驱动方式的如压力驱动,有很多的优点:1)一般而言,电渗流的速度大小与管道或槽道的横向尺寸无关,易于控制;而压力流的速度除了与压力梯度有关之外,还与管道或槽道的横向尺寸有关,为了保持一定的流速,需要考虑双重因素;

2)电渗流在管道或槽道中的横向速度剖面几乎是平直的,

有“平流泵”之作用,这样的速度剖面有利于样品的分离,即使在通道内传输很长距离,样品的浓度带宽变化也很小,而压力驱动流将产生抛物线型速度剖面,沿通道横向的速度梯度比较大,不利于样品的高效分离;3)电渗流主要通过施加电压驱动带电流体,因此可以通过控制电压来控制流速,利用电压的切换可以在微通道的交叉口控制电渗流的方向,实现阀的功能。优化通道的几何结构,还可以在微流装置的不同部位产生不同的流速。这在生化分析中,例如液体的混合及多样品的并行处理中很有用处。对于压力等驱动方式,通常需要安装微泵、微阀等装置,在工艺加工及维修方面比较困难。
当然,电渗流的驱动与控制也存在着一些缺点:1)电渗流对管道或槽道壁面材料和被驱动流体的物理化学性质有要求,与液体接触的表面材料必须能够提供电荷,以形成双电层,因此它只适合一定范围内的流体和管壁材料;2)产生电渗流所需要的高电压电源会带来安全、功耗和所占空间大的问题,这不利于系统的微型化;3)电渗流尽管适合于驱动和控制狭窄管道或槽道的微量液体,但由于焦耳热问题,它却不能高速驱动更宽管道中的流体,而这一能力在许多微流体应用(例如样品的预处理)中是有必要的。
1.2.2 芯片上阵列电极方式的电驱动 外置电极系统产生相应的电渗流需要高压,高压电源存在安全跟体积庞大的问题,随着MEMS技术的发展,能够在微流控芯片上集成阵列电极,考虑采用阵列电极来减低微流控芯片分析系统的操作电压,使之更有利于生化样品体系的检测。
(1)基于阵列电极的直流电驱动
徐溢 wwwhxtborg 《化学通报》在线预览版 微流控芯片中的流体驱动和
在电泳分离的过程中,施加高电压可以提高分离速度和分离柱效,改善分离性能。(www.61k.com)因此,现有的电泳芯片的研究多集中于将较高的分离电压施加在较短的分离通道上以获得很好的分离效果,但是较高的分离电压制约了电泳芯片向集成化、便携式等方向发展。为了解决高压电源对芯片实验室的束缚,提出了低压方式驱动下的芯片电泳。低压电泳芯片的基本思想是用阵列电极将分离管道分成许多小的分离区带,在一个或者几个区带之间施加电压,这样可以在使用一个较低电压的情况下获得原来的场强(如图3)。在施加了电压的电极对之间的区带,样品受到驱动并逐渐分离,转换电极以在分离区带产生必需的电场强度使样品得到连续的分离。
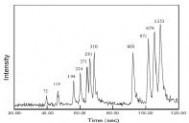
Lin等[18,19]介绍了低压电泳芯片的设计以及操作原理,并用有限元分析方法来模拟单边电极、双边电极以及组合电极三种不同类型的阵列电极的电泳芯片的电场特性。双边电极比起单边电极在管道中产生一个更加均匀的电场,组合电极采用了单边电极的设计但是能够提供一个更均匀和更加高强度的电场。同时,也考察了不同电极间距以及电极宽度下电场的分布情况以及电压电泳芯片电极设计的参数。Fu等[20]也做了低压电泳芯片方面的相关研究,对比了常规电压施加方式和低电压施加方式下的电势分布,模拟了不同方式下的样品节形状和样品分离谱图(图4),发现双边低压的电压施加方式能够得到形状较好的样品节,并且样品的分离谱图与常规施加方式下的样品谱图相对吻合,而单边施加方式下的样品节倾斜严重,样品分离谱图中样品的峰扩展严重,而且分离效果较差。温志渝等[21,22]进行了一系列低电压集成电泳芯片的研究,首先利用在分离通道上分段、交替、循环施加电压的方法,提出了电泳芯片低电压分离的模型,进而等根据电泳芯片模型的集成化分离过程,进行控制电路的设计和制作,利用多路开关原理来控制电极阵列中的对应电极的通断,从而在不同的时刻将分离电压施加在对应的一对电极上形成所需的分离场强。刘岗等[23]运用简化的低电压电泳芯片的运动梯度场的分离和控制模型,对低电压芯片的各参数与分离度、分离效率的关系进行了计算机模拟和讨论,提出低电压分离的电泳芯片的分离效率和分离度主要与控制次数、循环次数、分离电压、微电极阵列数、微电极间距以及被分离物质的淌度有关。陈里铭等[24]通过建立数学模型,对设置了阵列电极的分离管道上的电势分布进行模拟,并且与常规的芯片分离管道的电势分布进行了对比。陈超等[25,26]在Lin等提出用移动电场控制DNA分离的设计思想的基础上,根据理论模拟计算,设计了线性分布式的电极阵列,介绍了以普通载玻片和PDMS为基本材料的芯片制作工艺,并相应开发了一套微机控制系统,以精确控制反应中的电场的空间、时间分布,从而实时、全程控制电泳过程,并利用自制的电泳芯片,进行了进样实验,通道流体特性实验以及DNA分离初步实验,在通道流体特性试验中成功看到了与理论很相符的进样图谱,得到了初步的DNA电泳分离的荧光显微镜照片。
61阅读提醒您本文地址:

至今关于低电压电泳芯片的研究,较多还处于模拟阶段,只有极少量的验证性实验。而在实际的样品

中以上的电势分布及样品节浓度分布均是模拟的结果。在实际的样品分离过程中,有许多的因素将会影响
徐溢 wwwhxtborg 《化学通报》在线预览版 微流控芯片中的流体驱动和
实际的分离效果。(www.61k.com)应考虑的因素包括:1)在高电压施加方式下,管道内流体流动主要是受电渗流的驱动,而在低电压施加方式下,由于施加电压的范围比较窄,流体运动的驱动力是否仍然是电渗作用的结果,还是电泳作用为主导;2)由于低压电泳芯片的材料多为硅材料,硅的表面性质是否会对管道内的流体有很大的影响;3)电极的切换方式;4)分离不同样品时分离条件的选择;5)检测器的选择。
(2)基于阵列电极的交流电驱动
微流控芯片系统用于细胞介质是近年来该技术发展的一个热点。在芯片中对细胞实行操纵和驱动的重要模式是电场力和压力[27,28],目前基于介电电泳的细胞操纵和驱动模式等备受关注。Arnold等[29]利用介电电泳进行了细胞的隔离和培养,通过在电极阵列上形成一个热对流细胞对定位和培育细菌产生帮助。图5及图6为li等[30]应用十字交叉阵列电极,利用活细胞和死细胞之间的介电性质不同对活的和热处理后的李斯特菌进行了富集和分离,并获得了极高的分离效率。Ramadana等[31]利用电泳和介电电泳技术实现了微流控芯片上细胞和微球的连续诱捕和溶解,利用堡式阵列电极可以进行DNA样品的的预处理。



交流电渗驱动现象是采用电极阵列在交流电作用下的电渗流引起的。当电压作用于电极时,在电场作用下电荷将会集聚在电极表面并且形成双电子层,极化的电极与双电子层和电场的切向分量相互作用,这将在双电子层上产生作用力,从而使流体运动。Ramo等[32]的实验中,利用非对称电极交流电渗驱动液体,在以玻璃为基底材料的芯片上,有50个电极对,大小电极的电极宽度分别为100μm和10μm,间隙为10μm,电极对之间的距离为100μm,大小电极电极长均为20mm,其中每一大小电极的组合被称为一个电极对。张善亮等[33]做了微流道交流电水力泵的数值模拟及优化,对流道长度为5mm,宽0.4mm,厚度0.4mm的电水力泵做了优化设计的数值模拟,最后得到驱动电极宽度为0.01mm,电极间距为0.02mm。
此外还有在微流控芯片上集成阵列电极,在微阵列电极上施加脉冲电压作为驱动力来实现液滴传输、合并和拆分等操作[34]的报道。随着MEMS技术的发展,这类技术目前正以强劲势头向微流控芯片分析系统中渗透,成为关注的热点。
1.3 其他驱动方式
电水力驱(Electrohydrodynamic, EHD)动和电渗驱动都是由电场和流体中电荷的相互作用来产生驱动力的,但EHD需要在流体或流体-固体界面诱导产生自由电荷,通过电场与自由电荷的相互作用来驱动流体,它一般适用于导电率极低的液体。EHD驱动技术有多种形式,但其中两种最具代表性。一种是Bart等
[35]在前人研究的基础上提出的电势行波驱动的EHD诱导泵,它的驱动力主要由在流体-流体或流体-固体界面上诱导的自由电荷产生的。两种材料的介电常数或导电率不同,在电极阵列上施加电压就可以在材料界面诱导自由电荷产生,但由于电力是垂直作用在界面层的,所以直流电场或静态交流电场并不能产生驱
徐溢 wwwhxtborg 《化学通报》在线预览版 微流控芯片中的流体驱动和
动力,如果在电极阵列上施加一个电势行波,下面的材料界面就会产生预制同步运动的诱导电荷,由于材料的电荷松弛会使自由电荷的运动滞后于电势行波的运动,这样导致的电势行波与被诱导电荷之间的位移就会产生一个作用在界面上的电表面应力,从而驱动流体运动。(www.61k.com)另一种是Richter等[36]提出的直流电压驱动的EHD注射泵,它的驱动力为作用在流体离子上的库仑力,这些离子是通过电化学反应由电极注射进流体中的,电极需要与流体直接接触,这样在电场作用下,发射电极和接受电极之间就会产生一压力梯度,从而使得流体在两电极间流动。
从原理上讲,如果能够在固-液界面产生某种特定的表面张力梯度,就可以驱使液体在特定的方向流动,产生这种表面张力梯度的方法有两类:一类是通过改变固体支持面的润湿性[37],另一类产生表面张力梯度的方法是通过改变液体的成分或温度梯度来实现的[38]。
Mandou等 [39]利用离心力来进行微流体的驱动和控制,在他们提出的“Lab CD”系统中,采用光刻和模塑成型的方法在塑料圆盘上制作微管道网络,流体被装载在靠近圆盘中心的供液池中,当圆盘由马达带动旋转时,流体就在离心力的作用下沿着微管道网络向远离圆心的方向运动,流体速度的大小可以通过调节马达转速来控制,而且通过控制转速,管道在盘片上的分布和几何构型可以实现流体的混合和被动阀的功能。
61阅读提醒您本文地址:
Johann等[40]在微芯片上利用电渗流和压力的共同作用来驱动流体,进行粒子的分离和筛选。他们提出了一种新颖的管道设计,在这种管道设计下,通过一个简单的电源就可以进行电压的设定以及进行电转换,使得流量可以转换,以进行粒子的筛选。其中的压力驱动是通过在芯片上以一定的方式在储液池上设置差异而产生的。基于三T型管道的设计,控制从侧面管道引入的缓冲溶液的反面水压流来消除后压流干扰电动流和粒子分离过程。
关艳霞等[41]报道了一种简单、造价低、体积小、流速稳定、可长时间连续使用及流速易于调节的微泵的研制。它以吸水膜的毛细作用和大气蒸发相结合为驱动力,由储液管、蓄水池、吸水膜和蒸发孔组成。此微泵具有不外接能源,运行时间不受限制,任意长(环境的相对湿度<100%)的优点,而且采用低成本的传统加工制作技术,就可根据需要设计出不同流速的微型泵。
重力作为流体驱动力的方法,在常规的流动分析系统中早有应用。在芯片上采用重力驱动的优点是不需要额外的驱动力源和驱动装置,造价低廉,使用方便,可显著提高整体系统的集成度。目前多用于芯片上连续流动分析体系。陈宏等[42]将微流控芯片多相层流分离技术与离子选择性电极检测技术联用,利用重力驱动的芯片多相层流分离系统,在线净化生物试样。但这些文献报道的系统均采用手工进样方式[43,44],不具有连续自动换样功能,换样操作繁琐费时,效率低,影响了系统对不同试样的分析通量和实用性。为了解决上述问题,黄艳贞等[45]建立了一种重力驱动的可连续换样的微流控芯片流动分析系统,采用新型套管型换样接口可实现连续高通量的试样引入,采样频率达80~100样/h。在该系统中,采用在微通道出口处加装引流管提高流速。此外,采用了水平通道储液池,可在较长的时间内保持流速的稳定。也有人做了一些微管道中重力驱动方式下的理论分析[46,47],发现高度越低,重力的作用就越来越不明显。
2.结论
微流控芯片上的流体的驱动和控制技术很多,而且正在不断发展中。微机械泵由于包含微型可动部件,所能提供的压力非常有限,且制作工艺复杂、价格昂贵,使应用受限,而现在发展迅速的无阀微泵由于其结构较简单,在集成到微流控芯片上有很大的潜力,是当前的研究热点,虽然已经取得很大的进展,但是
徐溢 wwwhxtborg 《化学通报》在线预览版 微流控芯片中的流体驱动和
还未真正的进入市场。(www.61k.com)电渗驱动仍然是最常用的方式,控制方法简单,容易在微流控芯片分析系统中管道中应用,但是由于电渗流对管壁材料和被驱动流体的物理化学性质敏感,它只适用于一定范围的流体和管壁材料,同时产生电渗流需要高压电源,不利于系统的微型化,在微流控芯片上集成阵列电极能够解决高压问题,能够在较低的电压下进行生物样品的分离、微流体的驱动等操作,因此,微流控芯片使用低电压,有利于系统的集成化以及更多生物样品的分析,也使得以阵列电极为基础的低电压驱动方式得到越来越多的关注。其他的各种微流体驱动方式各有其优点,但是也存在一些局限性。
总之,微流体驱动技术是微流控芯片中的关键技术之一,由于流动尺度的减小,微流体的流动特性与宏观有很大的不同,许多在宏观尺度下可以忽略的现象在微观尺度下成为流体流动的主要影响因素。在微流体的驱动和控制技术中,微流体的特性的深入研究,有利于发展新的驱动技术以及改进已有的驱动和控制技术。
参考文献
[1] J G Smits. Sensors&Actuators, 1990, A21-23:203~206.
[2] H T G Van Lintel, F C M van de Pol, S Bouwstra. Sensors&Actuators, 1988, 15:153~167.
[3] E Stemme, G Stemme. Sensors&Actuators, 1993, A39:159~167.
[4] A Olsson, E Stemme. Sensors&Actuators A, 1995:549~556.
[5] R Zengerle, J Ulrich, S Kluge et al. Sensors&Actuators, 1995, A50:81~86.
[6] M Stehr, S Messner, H Sandmaier et al. In:Proc MEMS’96, San Diego, USA, 1996:485~490.
[7]尹执中, 庞江涛, 刘理天等. 清华大学学报(自然科学版), 2000, 40(6),36~38.
[8] N T Nguyen, T Q Truong. Sensors and Actuators B, 2004, 97 :137~143.
[9] T Q Truong, X Y Huang, N T Nguyen. Science, 2003, 4(2):249~252.
[10] D J Harrison, K Fluri, Z H Fan et al. Science, 1993, 261:895~897.
[11] D J Harrison, Z Fan, K Fluri et al. Science, 1994:21~24.
[12] R B M Schasfoort, S Schlautmann, J Hendrikse et al. Science, 1999, 286:942~945.
[13] F Bianchi, R Ferrigno, H H Girault. Analytical Chemistry, 2000, 72(9):1987~1993.
[14] L E Locascio, C E Perso, C S Lee. Journal of Chromatography A, 1999, 857:275~284.
[15] J L Pittman, C S Henry, S D Gilman. Analytical Chemistry, 2003, 75:361~370.
[16] Y Sun, C S Lim, A Q Liu, T C Ayi et al. Sensors and Actuators, 2007, A133: 340~348
[17] 姚波, 冯雪, 罗国安等. 高等学校化学学报, 2005, 26(1):43~45.
[18] Y Ch Lin, W D Wu. Sensors and Actuators B, 2001, 73:54~62.
[19] Y Ch Lin. Sensors and Actuators B, 2001, 80:33~40.
[20] L M Fu, R J Yang. Electrophoresis, 2003, 24:1253~1260
[21] 吴英, 温志渝, 张正元等.光学精密工程, 2003, 11(2):130~135.
[22] 李霞, 温志渝, 李星海等. 微纳电子技术, 2003, 7/8:344~346, 250.
[23] 刘岗, 温志渝, 李霞等. 微纳电子技术, 2003, 7/8:347~350.
[24] 陈里铭, 闫卫平, 刘军民. 2005, 26(8)增刊:183~184.
[25] 陈超, 赵湛, 张搏军. 微纳电子技术, 2003, 7/8:362~364.
[26] 陈超, 赵湛. 传感器技术, 2004, 23(1):77~80.
徐溢 wwwhxtborg 《化学通报》在线预览版 微流控芯片中的流体驱动和
[36] A Richer, A Plettner, K A Hofmann et al. Sensors&Actuators, 1991, A29:159~168.
[37] H Gau, S Herminghaus, P Lenz et al. Science, 1999, 283:41~42.
61阅读提醒您本文地址:
[38] B S Gallardo, V K Gupta, F D Eagerton et al. Science, 1999, 283:57~60.
[39] D C Mandou, G J Kellogg. SPIE, 1998, 3259:80~93.
[40] R Johann, P Renaud. Electrophoresis, 2004, 25:3720~3729.
[41]关艳霞, 戴敬, 方肇伦. 分析化学, 2005, 33(3):423~127.
[42]陈宏, 方群, 殷学锋, 方肇伦. 高等学校化学学报, 2004, 25(8):1428~1431.
[43] Wu X, Suzuki M, Sawada T et al. Analytical Science, 2000, 16:321~323.
[44] I B H Weig, R Bardell, T Schulte et al. Proceedings of MicroTAS[C], 2000:299~302.
[45] 黄艳贞, 方群, 李丹妮. 高等学校化学学报. 2004, 25 (9)1628~1631.
[46] Z X Cai, H W Chen, B Hen et al. Talanta 2006 (68) 895~901.
[47] W R Jong, T H Kuo, S W Ho et al. International Communications in Heat and Mass Transfer, 2007(34) 186~196.
徐溢
1966年9月生于重庆
2006年获重庆大学博士学位
现系重庆大学教授
主要从事分析化学、应用化学和微型集成生化分析系统等领域的研究 Email:xuyibbd@yahoo.com
61阅读提醒您本文地址:
二 : 化学通报在线预览版 14
http://www.hxtb.org 《化学通报》在线预览版
室温离子液体电解质与锂离子电池碳负极材料的相容性
郑洪河,石静,刘云伟,王键吉
(河南师范大学化学与环境科学学院 新乡 453007)
摘要 室温离子液体电解质与碳负极材料间的相容性是其应用于锂离子电池的关键问题之一。本文总结了室温离子液体电解质体系与碳负极材料相容性的研究现状和微观机制,阐述了不同种类的室温离子液体与碳负极材料相容性的规律和存在的问题以及改善方法。
关键词 锂离子电池 离子液体 碳负极 电解质 相容性
Compatibility of Ionic Liquid Electrolyte with Carbonaceous anodes
in Li-ion Batteries
Zheng Honghe, Shi Jing, Liu Yunwei, Wang Jianji
(College of Chemistry and Environmental Sciences, Henan Normal University, Xinxiang 453007)
Abstract Compatibility of ionic liquid electrolyte with carbonaceous anode is the key issue deciding the application of ionic electrolyte into lithium ion batteries. The recent status and micro-mechanisms relating to the compatibility of carbon with different ionic electrolytes were summarized. Some rules and problems relating to the compatibility are discussed. Different ways to improve the compatibility are proposed.
Keywords Li-ion batteries, Ionic liquid, Carbonaceous anode, Electrolyte, Compatibility
室温离子液体(简称RTIL) 具有导电性好、蒸汽压低、无可燃性、热容量大、电化学窗口宽和自放电少的优点,作为可替代挥发性有机溶剂的新型绿色溶剂引起了人们广泛的关注[1~4],近年来,用作
锂离子电池电解质在消除锂离子电池安全隐患方面也显示了良好的前景。但室温离子液体与电极材料间的相容性差,电极材料在离子液体电解质中难以表现出理想的脱嵌锂性能和循环性能,这是制约室温离子液体电解质用于锂离子电池的关键[5~7]。
本文探讨碳负极材笔者在前文[8]中详细论述了室温液体电解质与锂离子电池正极材料间的相容性,
料与各种室温离子液体电解质间的相容性。
________________________________________
国家自然科学基金(20573033)与河南省杰出青年基金(0412001100)资助
2007-10-17收稿,2008-02-01接受
1.室温离子液体电解质与碳负极材料相容性的主要问题
1.1锂离子传输问题
Lee等[9]研究了锂离子电池中阳离子的扩散情况。对比了1,2-二甲基-3-丁基咪唑六氟磷酸盐
其中几种离子在溶剂中的传输如图1所示,(BDMI-PF6)和LiPF6在溶剂聚碳酸酯(PC)中的离子传输机理。
PF6在三种混合溶剂中的扩散速度大体相同,但BDMI+在PC中的扩散系数比Li+高得多,在锂离子电-
池的充放电过程中,两种阳离子相互竞争,扩散较快的BDMI+先于Li+到达负极表面,并在负极表面还原分解,形成了一层具有保护作用的薄膜,阻碍了Li+在电极表面发生反应,致使电池难以表现出理想的性能。
BDMI
+
Li+ 图1两种阳离子在充电过程中竞争示意图[9]
Fig.1 Two Cations Competition during the Charge [9]
1.2离子液体的还原问题
离子液体的阳离锂离子电池的碳负极材料始终处于较低的电极电位条件(0.1V vs. Li+/Li)下, 因此,
子容易在碳负极表面发生还原反应而被分解,这种情况对于电化学窗口较窄的离子液体如咪唑类离子液体尤为明显。阳离子的还原反应不仅使电解质本身的性能下降,同时,也会破坏碳负极材料的结构和表面,因此,离子液体的电化学窗口也是决定其与碳负极材料相容性的重要指标。
1.3碳负极/离子液体间的界面问题
界面问题又包括界面成膜问题和界面浸润问题两个方面。在使用传统有机液体电解质的锂离子电池中,有机非质子溶剂在电池的首次充电过程中与碳负极发生反应,形成覆盖在碳电极表面的钝化薄层,人们称之为固体电解质中间相(Solid electrolyte interphase或简称SEI)膜[10]。优良的SEI膜具有有机溶剂不溶性,允许Li+较自由地进出电极而溶剂分子却无法穿越,从而阻止了溶剂分子共插时对电极的破坏,大大提高了电极循环寿命。室温离子液体电解质用于锂离子电池时,负极表面也同样需要一层薄而致密的保护膜,这个膜层应具有与在有机液体电解质中形成的SEI膜相同的性能,即能够阻止离子液体的阴阳离子嵌入石墨层间而允许Li+较自由地进出电极,这是实现碳负极材料与室温离子液体电解质相容性
的关键。
界面浸润问题则是源于室温离子液体与电极材料粘结剂如聚偏氟乙烯(PVDF)或聚四氟乙烯(PTFE)间的浸润性差,这使得电极材料与电解质间的界面阻抗特别高,比传统的有机液体电解质高5~10倍,从而大大影响电池的高倍率充放电性能。
2.不同室温离子液体电解质与碳负极材料间的相容性
2.1咪唑类室温离子液体电解质与碳负极材料间的相容性
咪唑类离子液体包括氯代-1-乙基-3-甲基-咪唑(EMI-AlCl3)、1-乙基-3-甲基-咪唑四氟硼酸盐(EMI-BF4)、1-乙基-3-甲基-咪唑三氟甲基磺酸亚胺盐(EMI-TFSI)等,具有粘度小、电导率高、与电极材料间的浸润性良好等优点,一度引起人们的广泛关注。其中N, N’-二烷基咪唑类离子液体在很大电势范围内是电化学惰性的[11],是目前所知综合性能最佳的离子液体。但这类离子液体电解质用于锂离子电池时的突出缺点是电化学窗口较窄(约4.2 V),石墨负极在其中的充放电效率较低,金属锂在其中的稳定性也较差。主要原因是由于金属锂表面不易生成具有保护性的膜层,也不能进行较为可逆的阴阳极极化。因而很多研究者只能选用锂合金或者金属氧化物作为负极材料。Fung等[12]以Li-Al合金作为电池的负极材料,检验了EMI-Cl的电化学性能。Nakagawa等[13]选择以Li4Ti5O12作为锂离子电池负极材料,这种电极能够有效避免充放电过程中EMI-BF4在阳极表面上的强烈的还原分解现象。 Garcia等[14]比较了EMI-BF4和EMI-TFSI的热稳定性、粘度、电导率、电化学稳定性以及电化学循环性能, 发现后者显示
了较优越的性能,Li4Ti5O12/LiTFSI+EMI-TFSI/LiCoO2电池经过200次循环后,可逆容量损失小于10%。
使用碳负极材料时,由于咪唑阳离子的扩散速度大于Li+,并且石墨负极的嵌脱锂电位低于咪唑阳离子的还原分解电位,电极在未达到嵌锂电位(0.7 V vs. Li/Li+)时,咪唑阳离子便在石墨表面剧烈还原分解(图2),阳离子的还原一方面破坏了电解质本身的性质,另一方面也在很大程度上破坏了石墨负极的表面,甚至是内部结构,所以要使Li+在石墨负极中进行可逆且有效的嵌脱锂循环,就必须设法阻止咪唑阳离子的还原分解。 2
Potential/Vvs.Li4+xTi5O121石墨电极的嵌锂电位0-1
0VVS.Li/Li+050100150200250300350400
?1Specific Charge /mAhg
图2 石墨电极在1M LiPF6/EMI-TFSI离子液体电解质中的首次恒电流充电曲线[15]
Fig.2 Electrochemical reduction curve for an SFG44 graphite electrode in 1M LiPF6/EMI-TFSI[15]
Ui等[16]考察了人造石墨、天然石墨、软炭和硬炭4种不同类型的碳为碳负极,在添加了少量SOCl2的LiCl型离子液体 氯代-1-乙基-3-甲基咪唑(AlCl3-EMIC)中的应用情况。在充放电循环实验中,4种碳负极的首次充放电效率均在40%~50%之间,这是由于,随着负极表面SEI膜的形成,SOCl2量随之减小。但是,经过30次循环后,这4种碳负极的放电容量分别为296、325、314和395 mAhg-1,且充放电效率分别达到93.2%、95.3%、90.0%和92.1%,显示出了优良的电极循环性能和很好的相容性。Holzapfel等[15,17]研究了人造石墨SFG44碳负极在1mol/L LiPF6/EMI-TFSI+碳酸乙烯酯(VC)(10(vol)%)电解液中的循环性能(图3),其可逆容量保持在350mAhg-1,循环100次后也观察不到明显的容量损失。扫描电镜实验显示,SFG44碳负极只有在添加VC后才能观察到SEI膜的形成,另外亚硫酸亚乙基酯(ethylene sulfite,ES)和丙烯腈也显示了较好的成膜作用,但是在碳负极表面的成膜效果均不如VC。总的来说,活性物质在含添加剂的室温离子液体电解液中比其在传统有机电解液中有着更好的稳定性。 Potential / V vs. Li4+xTi5O121电极钝化膜的形成电位00V vs. Li/Li+-1
-2-1 Specific Charge / mAhg
图3 石墨电极在添加10 (vol)% VC的1mol/L LiPF6/EMI-TFSI离子液体电解质中的首次恒电流充电曲线[15]
Fig.3 Fist electrochemical cycle for an SFG44 graphite electrode in 1mol/L LiPF6 /EMI-TFSI containing 10% vinylene carbonate[15]
2.2季铵盐类离子液体电解质
季铵盐类离子液体电解质与咪唑类离子液体电解质有所不同,季铵离子的离子半径一般都很大,并且由于体系的电化学窗口宽,阳离子的稳定性好,金属锂可以在其中稳定存在;但由于其稳定性好,在碳负极表面却不易发生还原分解反应,在电池首次充电过程中碳负极表面不能被有效钝化,季铵盐阳离子易先于Li+嵌入石墨层间,大体积阳离子的嵌入阻碍随后的Li+嵌入,因而碳负极在其中难以进行有效的嵌脱锂循环,季铵盐阳离子的嵌脱层成为电极反应的主要形式。Katayama等[18]研究了由三甲基己基
(TFSI-)组成的室温离子液体电解质TMHA-TFSI的循环伏安行为,铵阳离子(TMHA+)和磺酰亚胺阴离子
发现TMHA+先于Li+嵌入石墨层间,从而证实了这一结论。加入3mol/L 的成膜添加剂碳酸乙烯酯(EC)
后的TMHA-TFSI中的循环伏安行为表明,电解质中的Li+被EC优先溶剂化,并在电极首次充电过程中还原分解,电极表面先于TMHA+嵌层形成有效的SEI膜,表现出良好的循环性能。
研究了不同成膜添加剂对天然石墨电极在LiTFSI/TMHA-TFSIZheng等[19]使用天然石墨作负极材料,
中的电化学性质,发现天然石墨在该离子液体电解质中主要表现为TMHA+的嵌层反应,难以进行有效的可逆嵌脱锂反应。添加20 (vol)%的成膜添加剂Cl-EC后,天然石墨负极在其中伏安行为如图4[19]所示。在阴极过程中1.4 V时出现一微弱的还原峰,这是Cl-EC在石墨电极界面的还原分解反应,其分解产物沉积在电极表面,形成石墨/电解质相界面SEI膜,SEI膜的形成有效地抑制了TMHA+的嵌层,0.5 V处的 TMHA+嵌层反应不再出现, 而0 V附近锂离子嵌层反应的特征峰和随后阳极过程中0.2 V左右的脱锂峰的强度都明显增强,说明Cl-EC的加入显著改善了石墨电极的嵌脱锂性质,这种添加剂优于其它常用的VC、ES、EC等。电极的形貌分析表明,薄、均匀而又密实的SEI膜的形成是天然石墨在添加Cl-EC后的该室温离子液体电解质中表现出优良电化学性能的原因。
0.25
0.20
0.15
0.10
0.05
0.00
-0.05
-0.10
-0.15
-0.20
-0.25
-0.30Current density/mA+Potential/V vs.Li/Li
图4 天然石墨在含有20% Cl -EC 的1 mol/L LiTFSI/TMHA-TFSI中的伏安行为[19]
[19]Fig. 4 The Cyclic voltammogram of Graphite in 20% Cl-EC added 1 mol/L LiTFSI/TMHA-TFSI
2.3吡啶和哌啶类离子液体电解质
吡啶和哌啶类离子液体在结构和理化性质方面均与季铵盐类室温离子液体相似,可以看作具有环状取代基的季铵盐。目前常见的这两类离子液体的典型例子分别为1,2-二甲基-4-氟吡啶四氟化硼盐(DMFP-BF4)和N-甲基-N-丙(丁)基哌啶磺酸亚胺盐(PP13(4)-TFSI)。同季铵盐一样,这两类离子液体的电化学窗口也很宽(一般>4V),热稳定性也不错。特别是PP13(4)-TFSI ,电化学稳定窗口可以达到5.8V,明显大于BMI-PF6(4.7V) 和BMI-BF4(4.6V),并且PP13(4)-TFSI负极极限电位低(-0.3V vs. Li/Li+),更适合用作以锂为负极材料的锂离子二次电池的电解质[20]。
以吡啶阳离子为基础的DMFP-BF4作为锂离子电池的电解液,热稳定温度在300℃,并且可以在
化学通报在线预览版 14_化学通报
很宽的温度范围内和锂稳定共存,其电化学窗口约4.1V,氧化电位大于5V (vs. Li/Li+) [21]。Koch等[22]利用电化学阻抗法对金属锂在DMFP-BF4电解液中的稳定性进行了研究。结果表明,离子液体DMFP-BF4的还原产物在锂电极表面上无法形成致密的钝化膜,不能完全隔绝锂电极与本体电解液的接触,从而存在着一定的不可逆容量。
Sakaebe等[23~25]的研究指出,以PP13-TFSI为电解质的Li/LiCoO2电池可以表现出优良的性能。几种不同的脂肪族阳离子和氨基阴离子组成的几种不同的离子液体用作电解质时Li/LiCoO2电池的电学性能的研究表明,阴离子的结构和种类以及阳离子的种类大大影响离子液体的粘度,离子传导率,电化学稳定性等性能,因而进一步影响了其在Li/LiCoO2电池中的充放电性能。研究还表明,这类离子液体不仅具有很好的抗氧化性,并且和LiCoO2正极材料具有很好的相容性。
当然,仅仅和正极材料有很好的相容性是不够的,PP13-TFSI与金属锂和石墨-锂合金(C6Li)等负极材料同样显示出了很好的相容性。
Xu等[26]比较了在加入和未加添加剂的0.4M LiTFSI/PP13-TFSI电解质中金属锂在镍箔上的电化学沉积和溶出情况及其循环性能,其中加入5(wt)%EC可以大大提高Li+的脱嵌循环效率,10次循环后循环效率可以达到90~92%。
Lewandowski 等[27]也发现,加入一定的成膜添加剂后,金属锂和 C6Li负极都可以稳定存在于
此LiTFSI/PP13-TFSI电解质中, 特别是加入10(wt)%的VC后,C6Li电极的循环性能和库仑效率大大提高,
电极经过100次循环后仍可达到首次放电容量的90%,而添加EC则达不到这样的效果。
2.4 其它功能化室温离子液体与碳负极材料间的相容性
随着离子液体种类的迅速增加,人们已经开始根据不同电化学应用的需要,设计具有特殊官能团或者特殊阴离子的离子液体,以期改善其电化学性质,因而这些合成的功能化室温离子液体组装成的电池大多能够表现出理想的电化学性能。
Seki等[28]为了促进咪唑阳离子的电子离域作用,提高电池的安全性和可逆性,在其环上引进了一个给电子基团,制备出1,2-二甲基-3-丙基咪唑三氟甲基磺酰胺(DMPI-TFSI),并且与EMI-TFSI对比其在充放电实验中的性能。以EMI-TFSI和DMPI-TFSI分别装配的电池经过50次循环后,库仑效率都分别维持在一个很高的数值96%和99.5%。这些都更加进一步地证实,在咪唑环的2位上引进给电子基团,3位上引进较长的烷基基团,可以在一定程度上提高电池的安全性和可逆性。
Ishikawa等[29]将双(氟磺酰胺)离子(FSI-)与EMI+和N-甲基-N-丙基吡啶阳离子(P13+)结合,制备出EMI-FSI和P13-FSI两种离子液体。由于FSI-的引入, EMI-FSI的粘度很低,在室温下一直呈液态,而
。并且,锂盐在EMI-FSI和P13-FSI中的溶解性都很好,利于在且具有很高的离子传导率(16.5mScm-1)
锂离子电池中应用。他们还对比了天然石墨在添加了0.8M LiTFSI的三种离子液体电解质体系
EMI-TFSI,EMI-FSI和P13-FSI中的循环伏安性质,其中,以LiTFSI/ EMI-FSI和LiTFSI/P13-FSI为电解质的体系都表现出了非常好的电化学可逆性,经过30次充放电后,石墨在LiTFSI/EMI-FSI电解液中的容量仍能保持在360mAhg-1,显示出了很好的相容性。可以看出FSI-的引入,使得石墨负极在不含任何添加剂的纯离子液体电解质中表现出稳定且可逆的充放电容量。
Sato等[30]研究了石墨电极在N,N-二乙基-N-甲基-N-(2-甲基乙酯基)季铵磺酸亚胺盐(DEME-TFSI)中的电化学性质,发现虽然这种电解质的电化学窗口宽(6V),但是用作锂离子电池电解质时,由于醚氧键的不稳定性,电解质在碳负极表面存在着严重的还原分解现象,添加EC或VC后,可以在石墨表面优先形成优良的SEI膜,阻止离子液体的分解,进而显示出良好的嵌脱锂性质。图5是石墨负极分别在不含任何添加剂和添加了10 (vol) % VC 的LiTFSI/DEME-TFSI中的充放电曲线[30],由图可见,石墨电极在其中可以具有超过350 mAh/g的可逆容量,但首次循环的不可逆效率较高。 3.0
2.5
+ 3.02.5
Potential/V vs.Li/Li2.01.51.00.5(a)1st.Potential/V vs.Li/Li+2.0 1.51.00.5(b)2nd.(b)3nd.(b)1st.(b)2nd.(b)3nd.
(a)1st.
0.0-1Specific capacity of graphite/mAhg0.0?1Specific capacity of graphite/mAh
图5 石墨负极在电解质 (a) 0.9M LiTFSI/DEME-TFSI,(b)添加10 (vol)%VC中的充放电曲线[30]
Fig.5 The discharge and charge curves of graphite in electrolyte (a) 0.9M LiTFSI/DEME-TFSI and (b) containing 10(vol)% VC[30]
3改善负极材料/室温离子液体相容性的方法
改善碳负极材料与室温离子液体的相容性需要从电解质体系和碳负极材料的优化两方面入手。
3.1 离子液体电解质体系的优化
3.1.1混合溶剂 溶剂是电解质的主体成分,改变溶剂的组成可以改善电解质的物理化学性质,从而改善电解质与碳负极材料间的相容性。混合溶剂的使用主要包括三种情况:离子液体/离子液体、离子液体/有机溶剂和离子液体/聚合物电解质。
Minato等[31]研究了氰基季铵盐类离子液体和咪唑类离子液体形成的共溶剂( co-solvent )在锂离子电池中的应用,发现三种含氰基的季铵盐类离子液体(如图6所示)在室温下的电导率都在10-4S·cm-1左右,无法达到正常电解液电导率的要求,但与EMI-TFSI混合后,其电导率可以提高到10-3S·cm-1,循环伏安表明氰基季胺盐[I]和[II]可以阻止EMI-TFSI在负极的分解,这种新型的室温离子液体可以为碳负极提供具有保护性的表面膜,显示了其对电池负极的稳定性。
图6 三种氰基季铵盐的分子结构示意图[32]
Fig.6 Chemical structures of three kinds of cyano-containing quaternary ammonium[32]
Chagnes等[32]将γ-丁内脂(γ-BL)与室温离子液体1-丁基-3-甲基-咪唑四氟硼酸(BMI-BF4)3:2(体积比)混合后作溶剂,LiBF4作溶质,研究了石墨和Li4Ti5O12 作负极在此电解质中的电化学性能,发现该电解质在1V左右就在石墨负极表面氧化分解,产物沉积在石墨表面,Li+难以在石墨电极表面进行有效的插层反应,嵌脱锂循环无法进行;而Li4Ti5O12 在其中的首次充放电容量达157 mAhg-1,经过19次循环后,容量损失也只有11%。
Shin等[33]考察了N-甲基N-丙基吡咯烷鎓二(三氟甲基磺酰亚胺)室温离子液体(PYR13TFSI) 与聚
该复合电解质合物电解质P(EO)20LiTFSI组成的复合电解质[P(EO)20LiTFSI+PYR13TFSI]的电化学性能,
在40℃的电导率为6×104 S cm1,LiFePO4在其中的首次放电容量达150 mAh/g, 经过240次循环后仍--
保持86%的可逆容量。这一结果表明室温离子液体与聚合物的复合电解质具有很好的研究与开发价值。
3.1.2混合锂盐电解质 随着人们对碳负极/电解质相界面的理解的逐步深入,越来越清楚地认识到锂盐电解质的作用,锂盐不仅为电解质提供锂源,而且对电极的表面钝化起着十分重要的作用,因而,混合锂盐电解质的优势越来越清晰地展现出来。有些锂盐如LiBOB本身就具有很好的成膜作用,对碳负极的钝化十分有利,电解质中添加少量LiBOB可以取代成膜添加剂,甚至表现出出人预料的结果。另外,LiBF4 与LiPF6的混合,甚至三元锂盐成分的混合都表现出比单一锂盐成分优良的与碳负极材料的相容性。
3.1.3添加剂 成膜添加剂在室温离子液体电解质中具有特别重要的意义,电化学窗口宽的离子液体电解质可以钝化碳负极表面,防止离子液体的阳离子嵌入石墨层间;电化学窗口窄的离子液体电解质还可以通过钝化碳负极表面,防止离子液体在电极表面的还原分解。从这个意义上讲,添加剂的优选和优化对改善离子液体电解质与碳负极间的相容性至关重要。然而,目前添加剂的种类和数量仍然局限于有机液体电解质的添加剂,适合离子液体电解质的添加剂还有待开发。
3.1.4 新型室温离子液体电解质的开发 新型室温离子液体,主要是锂离子液体的开发对锂离子电池具有更加重要的意义,因为这类锂离子液体用作锂离子电池电解质不存在另类阳离子与Li+的竞争和共嵌入的问题。Fujinami等[34]报道过类似锂离子液体的盐类,但没有介绍这类离子液体的电化学性质。Shobukawa等[35]详细研究了锂离子液体的制备方法以及Li+在其中独特的运动方式,他们将1,1,1,3,3,3-六氟-2-丙氧基(HFIP)或五氟苯氧基(PFP)和甲氧基-齐聚环氧乙烷配体与LiBH4发生取代反应,合成
LiHFIP-n和LiPFP-n (n=3,4,7.2),这种盐在室温下为无色透明状液体,且随着n值的增加,自扩散系数也从10-9增加到10-8cm2·s-1,低于典型的咪唑类和吡啶类离子液体的自扩散系数[36]。在这种离子液体中,
阴离子在电场的作用下运动,而Li+的移动则是从一个“分子笼”Li+居于阴离子所构成的“分子笼”的中央,
中移动到另一个“分子笼”中。遗憾的是,还没有这类离子液体与碳负极材料间的相容性研究的相关报道。
3.2负极材料的优化
用作锂离子电池碳负极材料的种类繁多,结构复杂,因此,通过优化材料的结构与表面性质来 改善其与室温离子液体电解质间的相容性的工作十分艰巨,相关研究还很少。但总体来看,碳材料的优化应注意以下几个方面:1) 提高材料的导电性,由于离子液体电解质中锂离子迁移性差,电极材料的导电性更加重要,从这个意义上讲,天然石墨的性质优于中间相石墨微球;2) 改善材料的表面状况,降低碳材料与电解质间的界面阻抗;3) 优化电极材料的制备工艺,如减少高分子粘结剂的用量、提高电极的孔率,从而可以改善离子液体与电极间的浸润性。
4 结语
室温离子液体电解质用于锂离子电池与碳负极材料的相容性还存在许多迫切需要解决的问题,但基于室温离子液体的电解质正在逐步显示其优势,室温离子液体电解质的进一步优化将成为开发安全、绿色锂离子电池的重要途径。
参考文献
[1] 刘卉,陶国宏,邵元华 等, 化学通报, 2004, 67(11):795~801.
[2] J S Wilkes. Green Chem., 2002, 4: 73~80.
[3] Y Hu, H Li, X Huang et al. Electrochem. Commun., 2004, 6(1):28~32.
[4] H Nakigawa, S Izuchi, K Kuwana et al. Electrochem Soc., 2003, 150(6): A605~A700.
[5] Y Katayama, MYukumoto, T.Miura. Elelctrochem. Solid-State Lett., 2003,6(5):A96.
[6] T Aoshima, K Okahara, C Kiyohara et al. Power Sources, 2001, 97~98: 377.
[7] M U e, M Takeda, T Takahashi et al. Electrochem. Solid State Lett., 2002, 5(6): A119~A121.
[8]郑洪河,刘云伟,曲群婷 等,化学通报,2007,70(11):834~839.
[9] S Y Lee, H H Yong, Y J Lee et al. Phys. Chem. B, 2005, 109: 13663~13667.
[10]徐仲榆,郑洪河. 电源技术,2000, 24(3): 171~177.
[11] H Ohno, M Yoshizawa. Solid State Ionics, 2002, 154:303~309.
[12] Y S Fung, R Q Zhou. Power Sources, 1999, 81: 891.
[13] H Nakagawa, S Lzuchi, K Kuwana et al. Electrochem. Soc., 2003, 150(6): A695~A700.
[14] B Garcia, S Lavallee, G. Perron et al. Electrochimica Acta., 2004, 49: 4583~4588.
[15] M Holzapfel, C Jost, A Prodi-Schwab et al. Carbon, 2005, 43:1488~1498.
[16] K Ui, T Minami et. al. J.Power Sources, 2005, 146: 698~702.
[17] M Holzapfel, C Jost, P Novak. Chem. Commun., 2004, 10: 2098
[18] Y Katayama, M Yukumoto, T Miura. Electrochem. Solid-State Lett., 2003, 6: A96.
[19] H Zheng, K Jiang, T. Abe et al. Carbon, 2006, 44:203
[20]许金强,杨军,努丽燕娜 等. 化学通报(?),2005, 63: 1733~1738.
[21] J O Bockirs, A Reddy, M Gambca-Aldeco. New York: Kluwer Academic Publishers, 2002.
[22] V R Koch, C Nanjundiah, G. B Appetecchi. Electrochem. Soc., 1995, 142(7):L116~L118.
[23] H Sakaebe, H Matsumoto. Electrochem. Commun., 2003, 5: 594~598.
[24] H Sakaebe, H Matsumoto, K. Tatsumi. Power Sources, 2005, 146: 693~697.
[25] H Matsumoto, H Sakaebe, K Tatsumi. J. Power Sources, 2005, 146: 45~50.
[26]J Xu, J Yang , Y NuLi et al. J. Power Sources, 2006,160: 621~626.
[27]A Lewandowski, A Swiderska-Mocek, J. Power Sources, 2007, in press.
[28]S Seki, Y Kobayashi, H Miyashiro et al. Phys. Chem. Lett., 2006, 110: 10228~10230.
[29] M Ishikawa, T Sugimoto, M Kikuta et al. J. Power Sources, 2006, 162: 658~662.
[30] T Sato, T Maruo, S Marukane et al. J. Power Sources, 2004, 138: 253~261
[31] M Egashira, M Nakagawa, I Watanabe et al. J. Power Sources, 2005, 146: 685~688.
[32] A Chagnes, M Diaw, B Carre et al. J. Power Sources, 2005, 145: 82~88
[33] J H Shin, W A Henderson, S Scaccia. J. Power Sources, 2005,143:236~242.
[34] T Fujinami, Y Buzoujima. J. Power Source, 2003, 119: 438
[35] H Shobukawa, H Tokuda,S Tabata et al. Electrochimica Acta, 2004, 50; 1~5.
[36] Y Katayama, M Yukumoto, T Miura, Electrochem. Solid-State Lett. 2003, 6: A96.
三 : 【文字+视频】教你在Mac上体验Windows 10技术预览版
Windows 10 技术预览版已经可以免费下载和试用,虽然你是 Mac 的用户,但这并不意味着你就不能体验 Windows 10。Windows 10 为桌面带来了像语音助手 Cortana 这样的整合,并改善了多任务体验等新功能。

本文将为你介绍在 Mac 上使用 VMware Fusion 安装 Windows 10 技术预览版的方法,资深用户可以略过本教程。详情可查看下面视频或文字说明。
初期步骤:
1.下载 Windows 10 技术预览版的 ISO 文件,并保存到桌面。
2.下载 VMware Fusion,安装 DMG 文件。
3.双击 VMware Fusion 应用,点击打开开始安装过程。
4.VMware 要求你输入管理员密码,输入后点击OK。
5.点击勾选 “I want to try VMware Fusion 7 for 30 days“,然后点击继续。
6.输入邮件地址,点击继续。
7.VMware 再次要求你输入管理员密码,输入后点击 OK。
8.取消选择 “Yes, I would like to help improve VMware Fusion.”,然后点击 Done。
在你的虚拟机上安装 Windows 10
1.选择 Install from disc or image,然后点击继续。
2.点击 Use another disc or disc image,然后找到刚刚下载好的 Windows 10 ISO 文件,点击打开和继续。
3.点击 Customize Settings 和 Save 储存.vmwarevm 文件到~/Documents/Virtual Machines 文件夹。
4.处理器和内存方面,你可以点击 Processors & Memory 来改变设置,如果你的 Mac 是 8 核处理器,这里建议选择 4 核。
5.对于内存,你可以使用推荐的配置。但如果你的 Mac 有 16GB 的内存,那么可以将内存设置为 4096。
6.硬盘方面,建议选择 60GB。
7.选择完上述配置后,点击右上角的 x 来关闭定制选项。
8.点击播放按钮开始 Windows 10 预览版的安装。
9.接下来就是等待 Windows 10 的常规安装了,等待重启后便可进入桌面。
安装 VMware Tools
这里距离完成 Windows 10 安装还有一件事情,这涉及到 VMware Tools 的安装。
1.安装 VMware Tools,点击虚拟机(Virtual Machine)-在菜单栏安装 VMware Tools。
2.会弹出一个对话框,点击安装。
3.打开 Windows Explorer,选择 DVD Drive,双击 setup64 文件。
4.在 User Account Control 点击 Yes。
5.在 VMware Tools 安装界面点击 Next。
6.选择 Typical(典型)安装并点击 Next。
7.点击开始 VMware Tools 的安装。
8.点击完成。
9.点击 Yes 重启系统。
10.你的虚拟机安装将会重启,Windows 10 也将会重启并准备好待使用。
以上就是在Mac上体验Windows 10技术预览版视频和文字,希望能对大家有所帮助!
本文标题:在线字体预览-wwwhxtborg 《化学通报》在线预览版 微流控芯片中的流体驱动和61阅读| 精彩专题| 最新文章| 热门文章| 苏ICP备13036349号-1